R code
library(tidyverse)
library(knitr)
library(DT)
library(plotly)library(tidyverse)
library(knitr)
library(DT)
library(plotly)Often in bioinformatics we are working on files that are to large to be opened in R or on your computer. In R the file size is capped at 5 MB. If we try to open a larger file the following message appears

We will use the Terminal window in the bottom left corner to explore large files to determine their structure. In this window we can use Unix commands if there are available on your computer (they are available on Posit Cloud and Unity). Note these are Unix and not R commands. In terminal we can use the Unix command head to look at the first 50 lines in the file 23andMe_complete.txt in the data directory.
head -25 data/23andMe_complete.tsvNotice that the first 14 lines contain information about the file and have hash tags #. rsid numbers can be linked to data in NCBI’s SNP database and other resources. To look at the last 25 lines use tail
tail -25 data/23andMe_complete.tsvWe can see that the mitochondrial DNA only has 1 allele since it is a haploid rather the diploid genome
It is important to understand data and objects types, particularly when we are importing data. R will try to try to make a best guess, but it is up to you to make sure the data read into R as you would like.
All the data files for a lab will be in your RCloud project data directory. To import a file you must include the correct location of the file relative to where your .Rmd file is located. Let’s try to read in a SNP genotype file from 23andMe. To read this into R using read_tsv (tab separated values) and ignore the comment lines containing #.
SNPs_23andMe <- read_tsv("data/23andMe_complete.tsv", comment = '#')Look in the Environment window at the SNPs_23andMe object. Notice that the columns names are rs548049170, 1, 69869 and TT. This is because there is a hash tag on the line with the column names (as seen above in terminal). Bummer but we can work with that while importing the file
SNPs_23andMe <- read_tsv("data/23andMe_complete.tsv", comment = '#', col_names = FALSE) |>
setNames(c("rsid","chromosome","position","genotype")) Look again in the Environment window at the SNPs_23andMe object. Notice the column names are now correct, but we want chromosome and `genotype to be treated as factors. Let’s make the change now.
SNPs_23andMe <- SNPs_23andMe |>
mutate(chromosome = as.factor(chromosome)) |>
mutate(genotype = as.factor(genotype)) Data transformation for making graphs and statistical analyses is a big part of this course. Today we will touch upon a few of the basics using the above data. Check out dplyr vignettes for more examples. Manipulating data in the tidyverse uses pipes |> or %>%to chain together steps. So if we want to filter theUMass_COVID_datato cases greater than 50. In this case we are not changing what is in theUMass_COVID_data` data object
SNPs_23andMe |>
filter(position < 10) # A tibble: 3 × 4
rsid chromosome position genotype
<chr> <fct> <dbl> <fct>
1 i4001200 MT 3 T
2 i4001110 MT 7 A
3 i4001358 MT 9 G We can string them together
SNPs_23andMe |>
filter(genotype == 'T') |>
filter(chromosome == 'MT') |>
filter(position < 100) # A tibble: 19 × 4
rsid chromosome position genotype
<chr> <fct> <dbl> <fct>
1 i4001200 MT 3 T
2 i4001079 MT 40 T
3 i4001177 MT 46 T
4 i4000979 MT 51 T
5 i3001827 MT 52 T
6 i3002101 MT 55 T
7 i702250 MT 55 T
8 i3002191 MT 57 T
9 i700868 MT 57 T
10 i3000534 MT 58 T
11 i706514 MT 58 T
12 i4001158 MT 59 T
13 i3000622 MT 60 T
14 i3001085 MT 63 T
15 i703460 MT 63 T
16 i3001577 MT 72 T
17 i704499 MT 72 T
18 i3001616 MT 89 T
19 i702733 MT 89 T To the number of rsids for each chromosome (notice we are not counting rsids but the number of times each factor in chromosome appears)
SNPs_23andMe |>
count(chromosome)# A tibble: 25 × 2
chromosome n
<fct> <int>
1 1 114988
2 10 73347
3 11 70624
4 12 67591
5 13 51454
6 14 44766
7 15 41849
8 16 44765
9 17 40972
10 18 40494
# ℹ 15 more rowsSNPs_23andMe |>
filter(chromosome == 'MT') |>
count(chromosome)# A tibble: 1 × 2
chromosome n
<fct> <int>
1 MT 5474We can arrange the order of the chromosomes by the number of rsids
SNPs_23andMe |>
count(chromosome) |>
arrange(n)# A tibble: 25 × 2
chromosome n
<fct> <int>
1 Y 4895
2 MT 5474
3 21 19379
4 22 20712
5 19 29536
6 20 34161
7 X 35928
8 18 40494
9 17 40972
10 15 41849
# ℹ 15 more rowsTo make a table of genotype counts for each chromosome use group_by
SNPs_23andMe |>
group_by(chromosome, genotype) |>
count(genotype)# A tibble: 340 × 3
# Groups: chromosome, genotype [340]
chromosome genotype n
<fct> <fct> <int>
1 1 -- 2223
2 1 AA 18904
3 1 AC 2221
4 1 AG 9941
5 1 AT 78
6 1 CC 24747
7 1 CG 131
8 1 CT 10113
9 1 DD 114
10 1 DI 6
# ℹ 330 more rowsTo select a subset of columns
SNPs_23andMe |>
select(rsid, chromosome)# A tibble: 1,418,387 × 2
rsid chromosome
<chr> <fct>
1 rs548049170 1
2 rs4477212 1
3 rs9326622 1
4 rs116587930 1
5 rs3094315 1
6 rs3131972 1
7 rs12184325 1
8 rs12567639 1
9 rs114525117 1
10 rs12124819 1
# ℹ 1,418,377 more rowsNote that none of the above creates a new data object. To do this and display
SNPs_23andMe_counts <-SNPs_23andMe |>
count(chromosome)
# to display data frame / tibble
SNPs_23andMe_counts# A tibble: 25 × 2
chromosome n
<fct> <int>
1 1 114988
2 10 73347
3 11 70624
4 12 67591
5 13 51454
6 14 44766
7 15 41849
8 16 44765
9 17 40972
10 18 40494
# ℹ 15 more rowsUse mutate to add a new column
SNPs_23andMe_summarize <-SNPs_23andMe |>
group_by(chromosome) |>
summarize(rsid_count = n()) |>
mutate(rsid_percent = rsid_count/sum(rsid_count))
# to display data frame / tibble
SNPs_23andMe_summarize# A tibble: 25 × 3
chromosome rsid_count rsid_percent
<fct> <int> <dbl>
1 1 114988 0.0811
2 10 73347 0.0517
3 11 70624 0.0498
4 12 67591 0.0477
5 13 51454 0.0363
6 14 44766 0.0316
7 15 41849 0.0295
8 16 44765 0.0316
9 17 40972 0.0289
10 18 40494 0.0285
# ℹ 15 more rowsThere are other functions you can use in summarize (https://dplyr.tidyverse.org/reference/summarise.html). For example, getting the minimum or maximum value in a range.
SNPs_23andMe |>
group_by(chromosome) |>
summarize(min_position = min(position), max_position = max(position)) # A tibble: 25 × 3
chromosome min_position max_position
<fct> <dbl> <dbl>
1 1 69869 249222527
2 10 95844 135508269
3 11 128623 134945120
4 12 68276 133838353
5 13 19025153 115106996
6 14 19000060 107287663
7 15 20004966 102462479
8 16 68031 90163275
9 17 1389 81162706
10 18 11358 78015180
# ℹ 15 more rowsYou can make tables using knitr
library(knitr)
kable(SNPs_23andMe_counts, caption = "Number of rsids on each chromosome")| chromosome | n |
|---|---|
| 1 | 114988 |
| 10 | 73347 |
| 11 | 70624 |
| 12 | 67591 |
| 13 | 51454 |
| 14 | 44766 |
| 15 | 41849 |
| 16 | 44765 |
| 17 | 40972 |
| 18 | 40494 |
| 19 | 29536 |
| 2 | 115061 |
| 20 | 34161 |
| 21 | 19379 |
| 22 | 20712 |
| 3 | 94371 |
| 4 | 83758 |
| 5 | 82777 |
| 6 | 92896 |
| 7 | 75442 |
| 8 | 71766 |
| 9 | 61381 |
| MT | 5474 |
| X | 35928 |
| Y | 4895 |
This works well for small tables, but for even moderate tables like above it takes up a lot of space. In the above example
A better option is using the DT package, but *** Don’t do this with tables of hundreds of thousands of rows (as in your complete SNP table).
library(DT)
datatable(SNPs_23andMe_counts)We can click on the table to reorganize the data.
Another good reference for ggplots is Cookbook for R by Winston Chang Please take your time and go through the following web pages.
Here are a couple of cheatsheets that can be useful
As of writing this lab I have not been able to control computed figure dimensions in a R chunk with Quarto executions, but the R Markdown format is working. The dimensions of an individual graph in a R Markdown document be adjusted by specifying the graph dimensions as below
# to adjust figure size {r, fig.width = 8, fig.height = 2}
ggplot(data = SNPs_23andMe, mapping = aes(x = genotype)) +
geom_bar() +
ggtitle("Total SNPs for each genotype") +
ylab("Total number of SNPs") +
xlab("Genotype")
# to adjust figure size {r, fig.width = 3, fig.height = 3}
ggplot(data = SNPs_23andMe, mapping = aes(x = genotype)) +
geom_bar() +
ggtitle("Total SNPs for each genotype") +
ylab("Total number of SNPs") +
xlab("Genotype")
You may have realized that you can export plots in R Studio by clicking on Export in the Plots window that appears after you make a graph. You can save as a pdf, svg, tiff, png, bmp, jpeg and eps. You can also write the output directly to a file. This is particularly useful for controling the final dimensions in a reproducible way and for manuscripts.
# Plot graph to a pdf outputfile
pdf("images/SNP_example_plot.pdf", width=6, height=3)
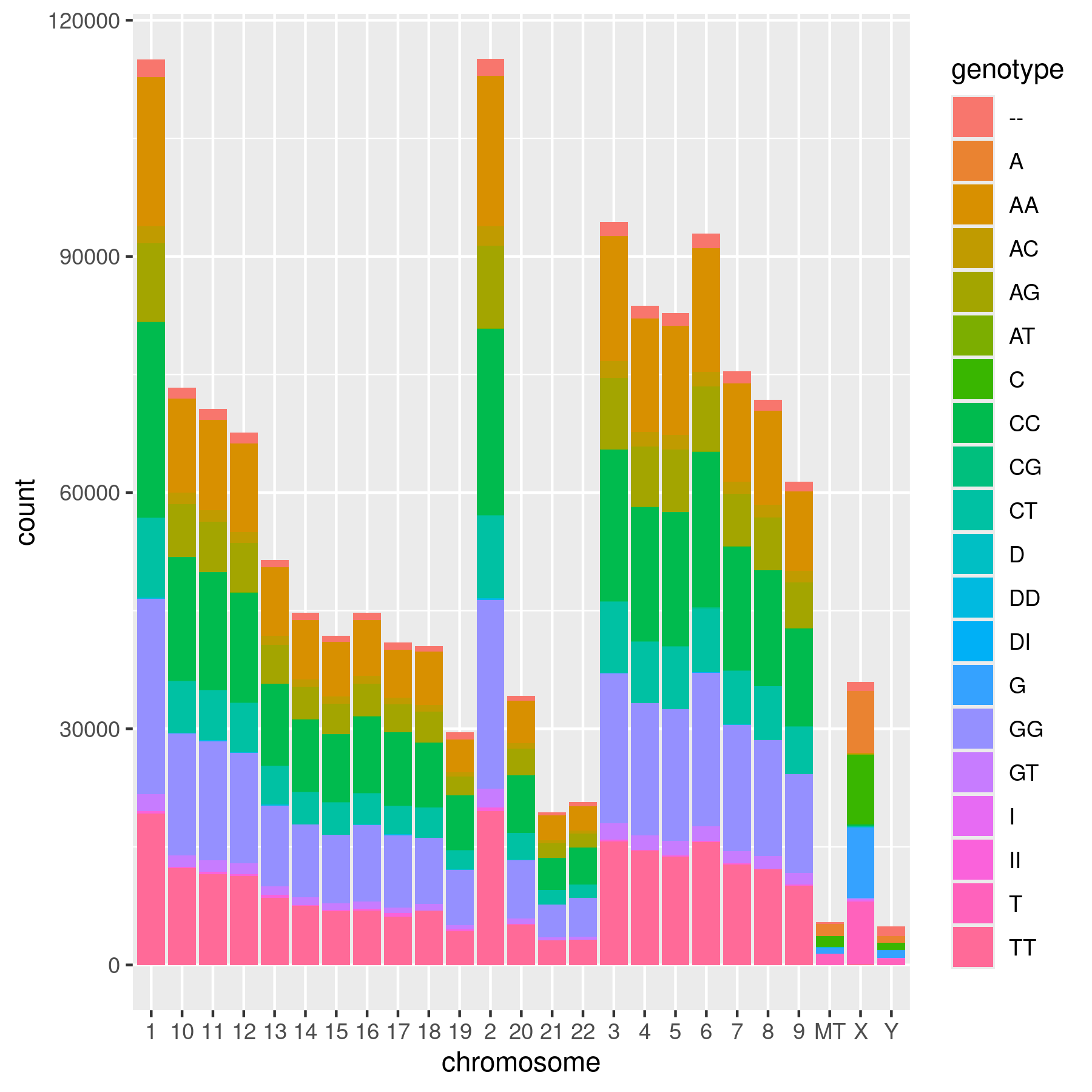
ggplot(data = SNPs_23andMe) +
geom_bar(mapping = aes(x = chromosome, fill = genotype))
dev.off()png
2 # Plot graph to a png outputfile
ppi <- 300
png("images/SNP_example_plot.png", width=6*ppi, height=6*ppi, res=ppi)
ggplot(data = SNPs_23andMe) +
geom_bar(mapping = aes(x = chromosome, fill = genotype))
dev.off()png
2 For more details on sizing output Cookbook for R - Output to a file - PDF, PNG, TIFF, SVG
Sometimes it is useful in controling the image layout for a report to file with the graph and then subsequently load it into the .Rmd file. This works with png files, but not pdfs. You can also upload images made with other bioinformatic tools into your RMarkdown report.
# This is the RMarkdown style for inserting images
# Your image must be in your working directory
# This command is put OUTSIDE the r code chunk
Another way to present a graph without the code is adding echo = FALSE within the r{} chunk - {r echo = FALSE}. This prevents code, but not the results from appearing in the knitr file.
With plotly/ggplotly (https://plot.ly/ggplot2/) you can make interactive graphs in your lab report.
library(plotly)# Version 1 1
p <- ggplot(data = iris, aes(x = Sepal.Length, y = Sepal.Width, color = Species)) +
geom_point()
ggplotly(p)# Version 2
ggplotly(
ggplot(data = iris, aes(x = Sepal.Length, y = Sepal.Width, color = Species)) +
geom_point()
)Use the 23andMe complete data set for the exercises. Pay attention to how your graphs look in today’s final knitted lab report. You will be docked points if the graphs do not look nice (e.g. overlapping column names, truncated legends, ets.)
Subset (select) the 23andMe complete data frame to just position and genotype
Filter to just the MT chromosome and remove genotypes A and T. (you can use != to remove).
Use group_by() and summarize() to find the first position, last position and number of positions (SNPs) measured for each chromosome. Note that each position represents a SNP, therefore the number of SNPs for a chromosome = the number of positions measured on each chromosome.
Building on ex3 create use mutate to create new column with the density of SNPs for each chromosome based the total number of SNPs divided by the last position - first position
Building on ex3 sort chromosomes based on SNP density.
Make a table for your report using DT to show SNP density
Using ggplot make a make a bar graph of the total SNP counts for each chromosome. Add title and labels for the x and y axis. Set the fill of the bars to yellow and the outline (color) to black.
Modify ex 7 to make a stacked bar graph with the contributions of each genotype to the total SNP count (Hint: use fill).
Turn ex 8 into an interactive graph using plotly. Note: Titles in plotly often need to be wrapped. I do this using labs(title = str_wrap("Total number of SNPs on each chromosome", 40).
Use facet_wrap to show each genotype as a separate graph (with a bar plot of counts per chromosomes).
Revise ex10 using scales="free_y" in facet_wrap to allow better show the variation in each graph.
Using the graph you made in exercise 8 output a PNG file with the ppi = 300 and the appropriate width and height so the image looks nice.
Load the png file of the graph from your images folder into your Quarto document.